Survival in PAH-CTD Compared With IPAH
In the REVEAL Registry, patients with pulmonary arterial hypertension associated with connective tissue disease (PAH-CTD) had significantly worse survival than patients with idiopathic PAH (IPAH).1*
RHC stratified by group 1 PH subgroup1
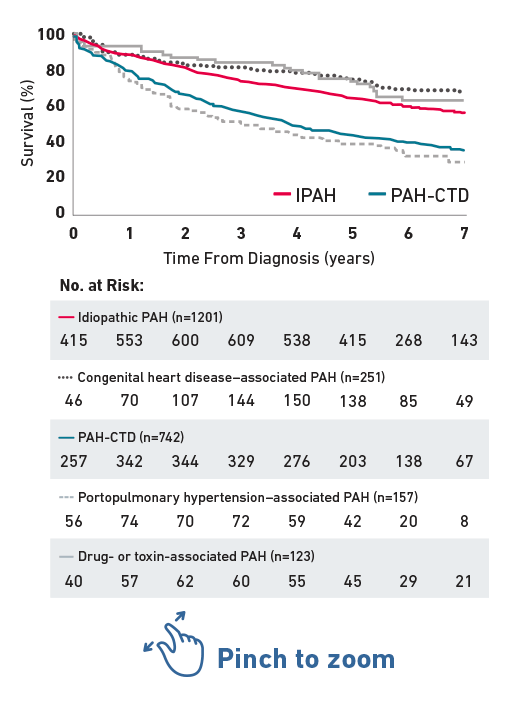
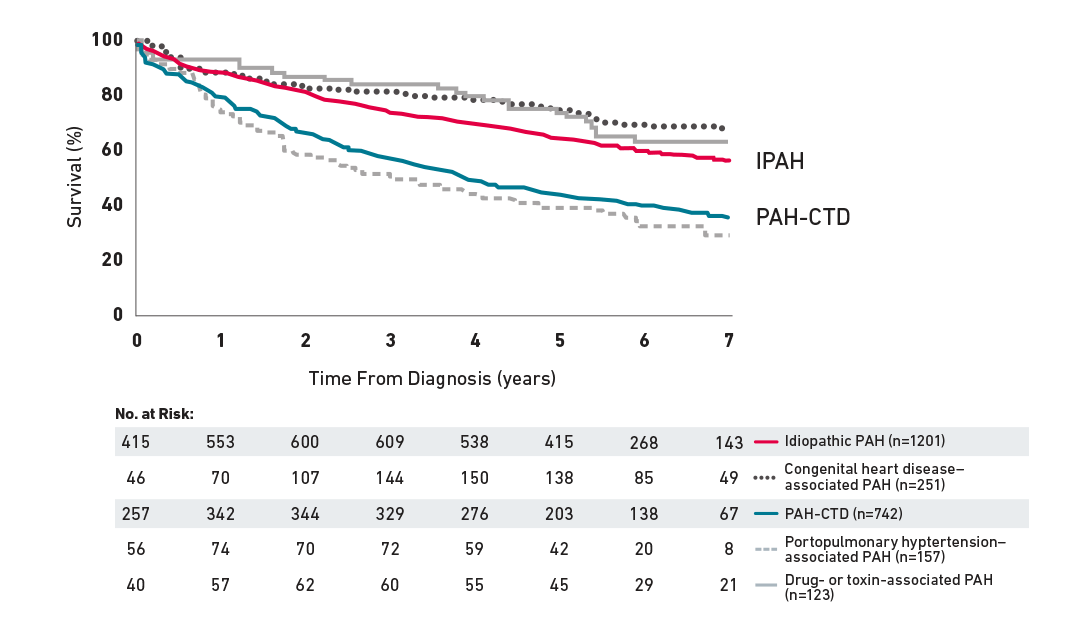
Reprinted from CHEST, Vol 142, Benza RL, et al, An Evaluation of Long-term Survival From Time of Diagnosis in Pulmonary Arterial Hypertension From the REVEAL Registry, pp448-456, 2012, with permission from Elsevier.
This observational, prospective REVEAL Registry study was conducted at 55 sites in the US, including 2635 patients who met the RHC criteria for PAH. Patients aged ≥3 months at diagnosis who met the modified definition for Group 1 PH were enrolled consecutively beginning in March 2006. Enrollment of previously diagnosed patients ended in September 2007, whereas enrollment of newly diagnosed patients continued through December 2009. Patients who were diagnosed before November 2001 were excluded from the analysis so that all analyzed patients were diagnosed within the contemporary treatment era, in which multiple approved classes of therapy were available.1
Among patients in the PHAROS Registry† with PAH associated with systemic sclerosis (SSc), or scleroderma, PAH WAS THE LEADING CAUSE OF DEATH WITHIN 4 YEARS OF PAH DIAGNOSIS.
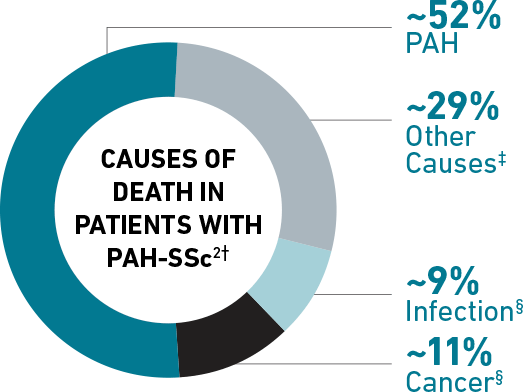
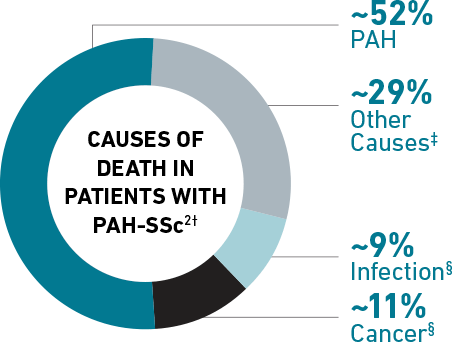
Shared pathophysiologic features between SSc and PAH may contribute to advanced presentation and poor prognosis at diagnosis.3-7 See the PAH-SSc Link >
Rapid Progression
PAH-SSc can progress quickly, drastically shortening the lives of patients:

In the REVEAL Registry,
49%
of patients with newly diagnosed PAH-SSc died within 3 years of PAH diagnosis.8*
Use minimally invasive tests to screen for possible PAH-SSc and the need for RHC.9 Learn More >
*In the REVEAL Registry, patients with new diagnoses were defined as those enrolled within 90 days of diagnostic RHC.8 REVEAL was funded and sponsored by Actelion Pharmaceuticals US, Inc.
†The PHAROS Registry was partially funded by Actelion Pharmaceuticals Ltd. Percentages do not add up to 100% due to rounding.
‡SSc-related and SSc-unrelated.
§SSc-unrelated.
IPAH=idiopathic PAH; PAH=pulmonary arterial hypertension; PAH-CTD=pulmonary arterial hypertension associated with CTD; PH=pulmonary hypertension; PHAROS=Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma; REVEAL=Registry to EValuate EArly and Long-term PAH Disease Management; RHC=right heart catheterization; SSc=systemic sclerosis.
References: 1. Benza RL, Miller DP, Barst RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from the REVEAL Registry. CHEST. 2012;142(2):448-456. 2. Kolstad KD, Li S, Steen V, et al; PHAROS Investigators. Long-term outcomes in systemic sclerosis-associated pulmonary arterial hypertension from the Pulmonary Hypertension Assessment and Recognition of Outcomes in Scleroderma Registry (PHAROS). CHEST. 2018;154(4):862-871. 3. Zanatta E, Polito P, Famoso G, et al. Pulmonary arterial hypertension in connective tissue disorders: pathophysiology and treatment. Exp Biol Med. 2019;244(2):120-131. 4. Chaisson NF, Hassoun PM. Systemic sclerosis-associated pulmonary arterial hypertension. CHEST. 2013;144(4):1346-1356. 5. Gaine S. Pulmonary hypertension. JAMA. 2000;284(24):3160-3168. 6. Condliffe R, Kiely DG, Peacock AJ, et al. Connective tissue disease–associated pulmonary arterial hypertension in the modern treatment era. Am J Respir Crit Care Med. 2009;179(2):151-157. 7. Vachiéry J-L, Coghlan G. Screening for pulmonary arterial hypertension in systemic sclerosis. Eur Respir Rev. 2009;18(113):162-169. 8. Chung L, Farber HW, Benza R, et al. Unique predictors of mortality in patients with pulmonary arterial hypertension associated with systemic sclerosis in the REVEAL registry. CHEST. 2014;146(6):1494-1504. 9. Khanna D, Gladue H, Channick R, et al. Recommendations for screening and detection of connective-tissue disease associated pulmonary arterial hypertension. Arthritis Rheum. 2013;65(12):3194-3201.